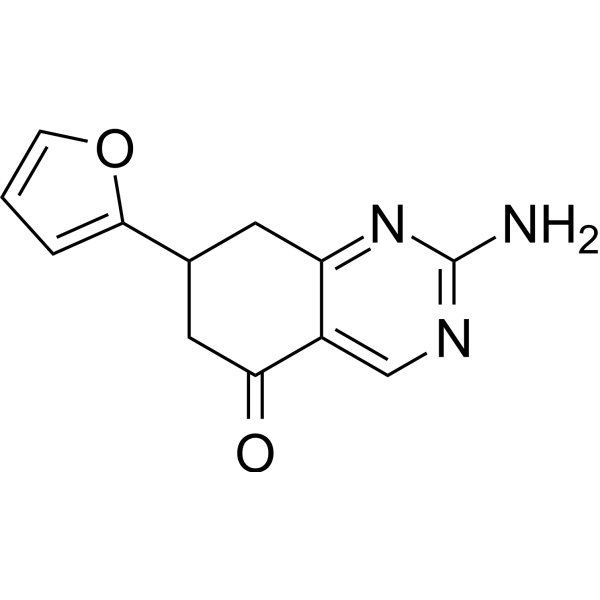
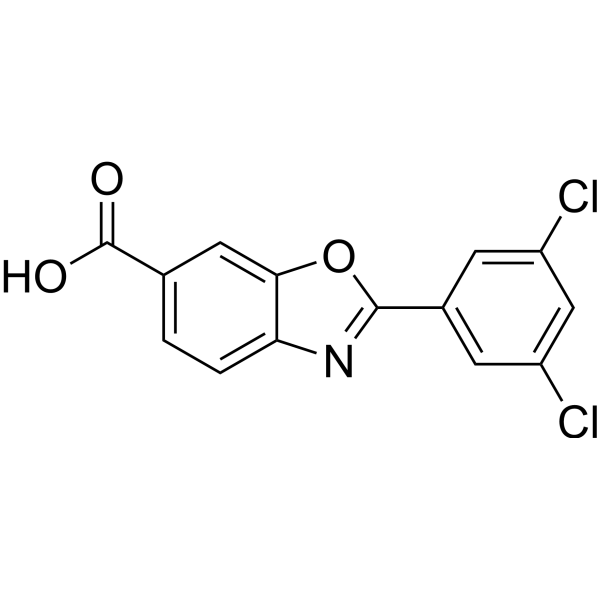
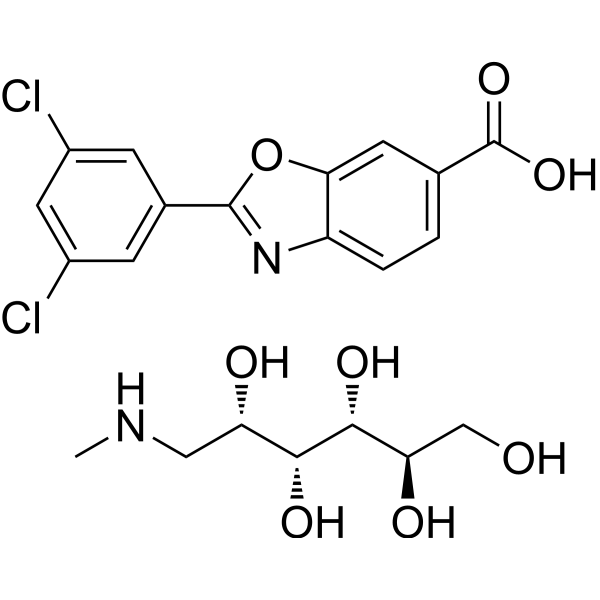
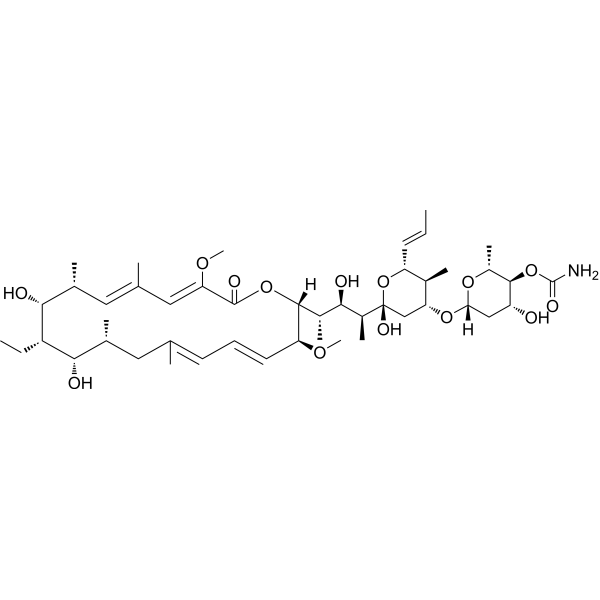
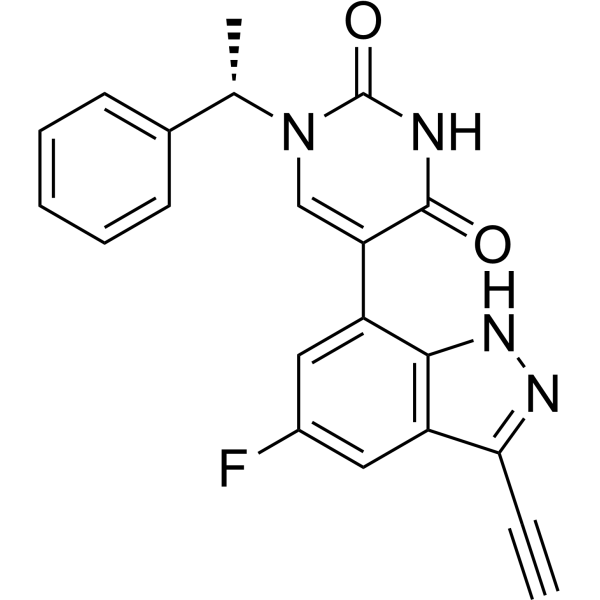
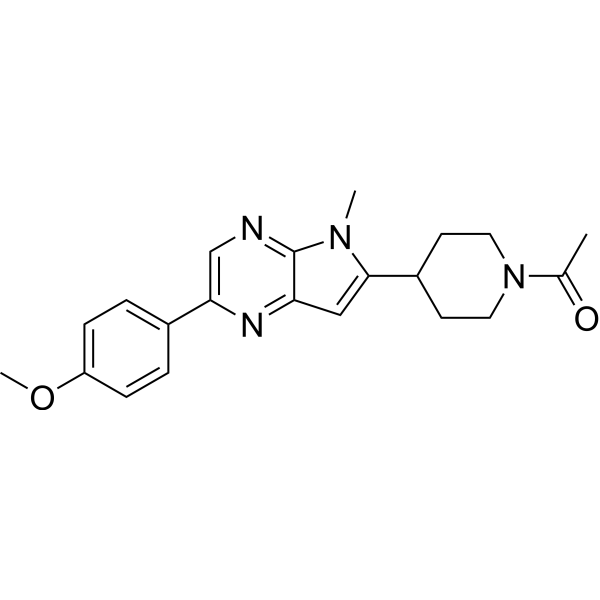
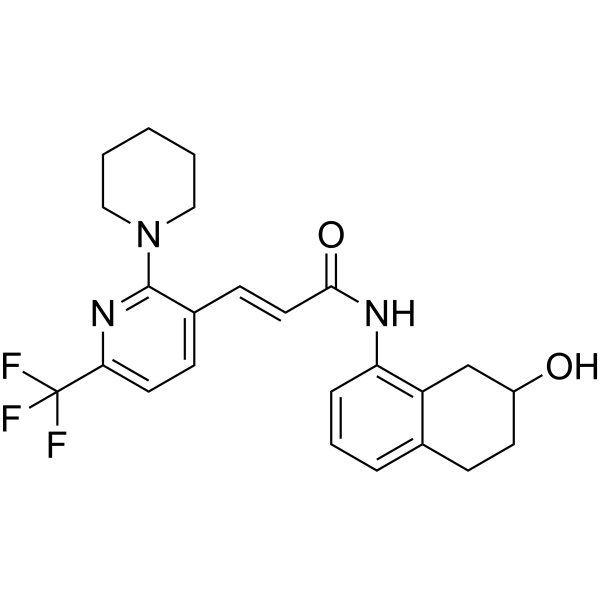
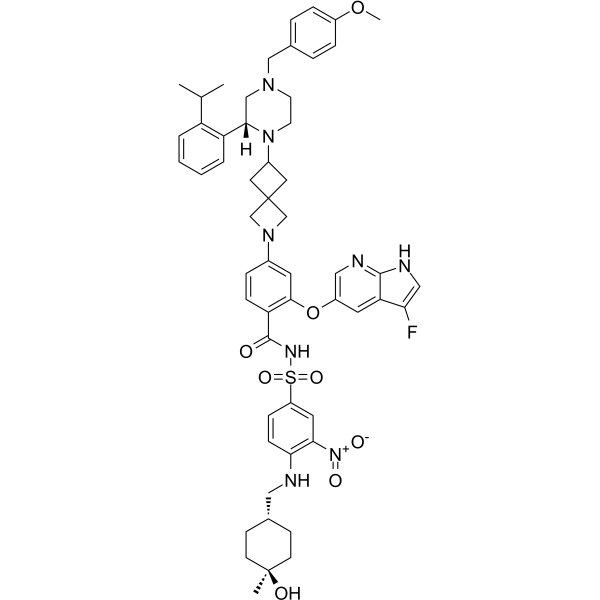
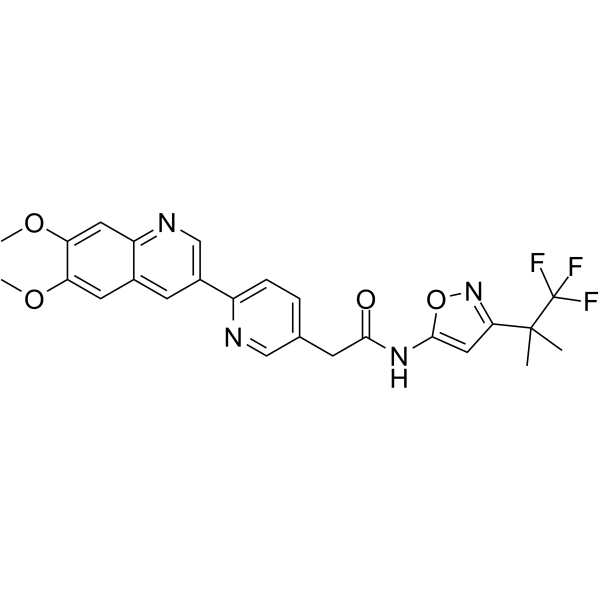
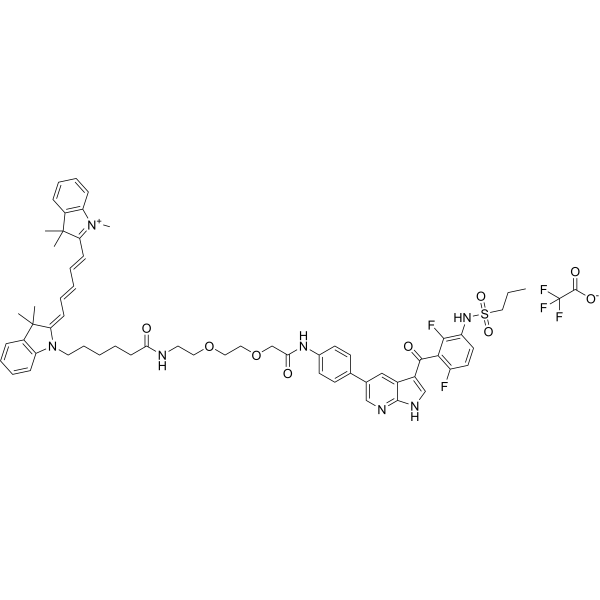
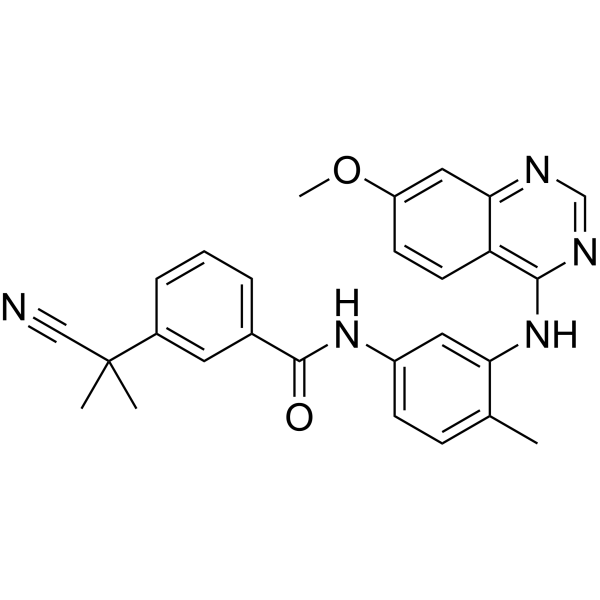
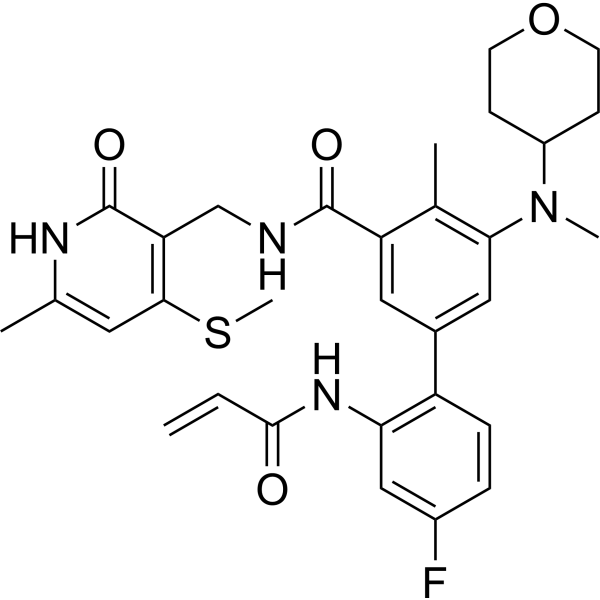
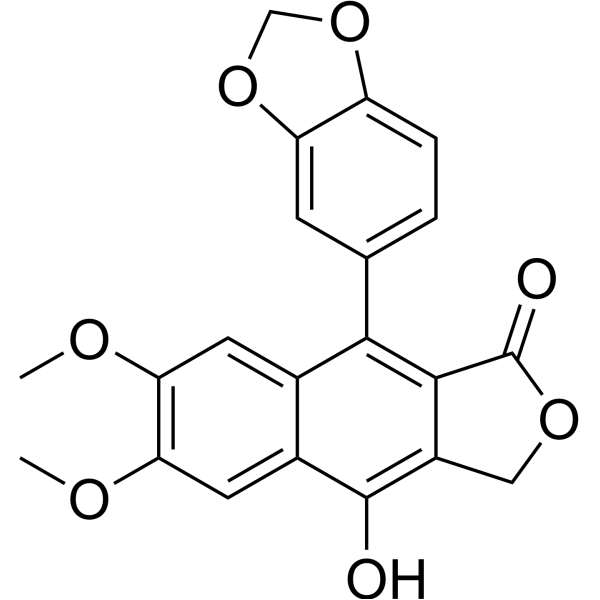
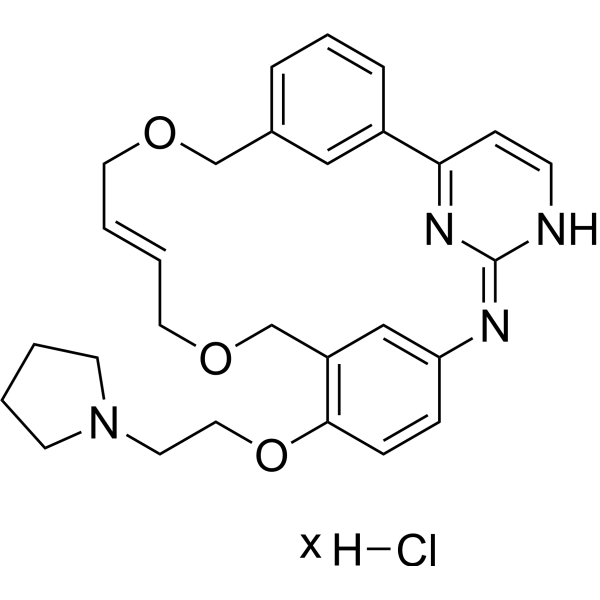
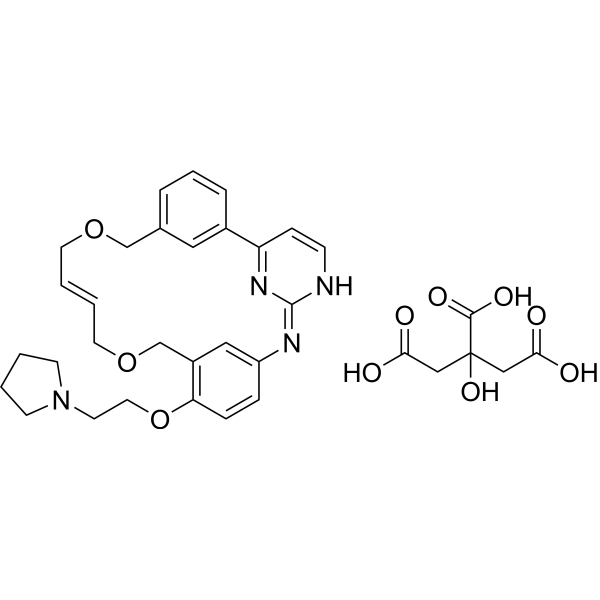
From 11:00 pm to 12:00 pm EST ( 8:00 pm to 9:00 pm PST ) on January 6th, the website will be under maintenance. We are sorry for the inconvenience. Please arrange your schedule properly.
NKY80 is a potent, selective and non-competitive adenylyl cyclase (AC)typeV isoform inhibitor with IC50s of 8.3 µM, 132 µM and 1.7 mM for typeV, III and II, respectively. NKY80 is a non-nucleoside quinazolinone and regulates the AC catalytic activity in heart and lung tissues .
Tafamidis is a potent and selective transthyretin (TTR) stabilizer, shows comparable potency and efficacy to the mutumant homotetramers V30M-TTR, V122I-TTR and wild type WT-TTR, with EC50s of 2.7-3.2 μM. Tafamidis inhibits amyloidogenesis .
Tafamidis meglumine (Fx-1006A) is a potent and selective transthyretin (TTR) stabilizer, shows comparable potency and efficacy to the mutumant homotetramers V30M-TTR, V122I-TTR and wild type WT-TTR, with EC50s of 2.7-3.2 μM. Tafamidis meglumine inhibits amyloidogenesis .
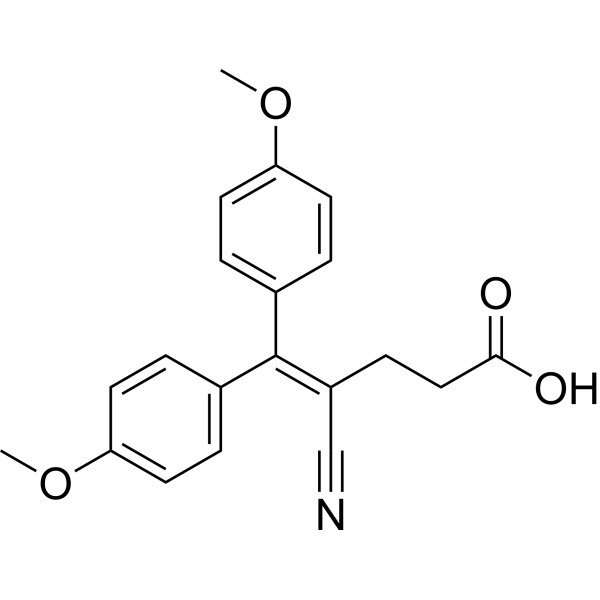
Satigrel (E5510) is a potent inhibitor of platelet aggregation. Satigrel inhibits collagen- and arachidonic acid-induced platelet aggregation through preventing thromboxane A2 synthesis by selective inhibition of the target enzyme, PGHS1, which exists in platelets. Satigrel inhibits PGHS1 (IC50: 0.081 μM) and PGHS2 (IC50: 5.9 μM). Satigrel is against Type III PDE, TypeV and Type II (IC50: 15.7 μM, 39.8 μM and 62.4 μM, respectively) .
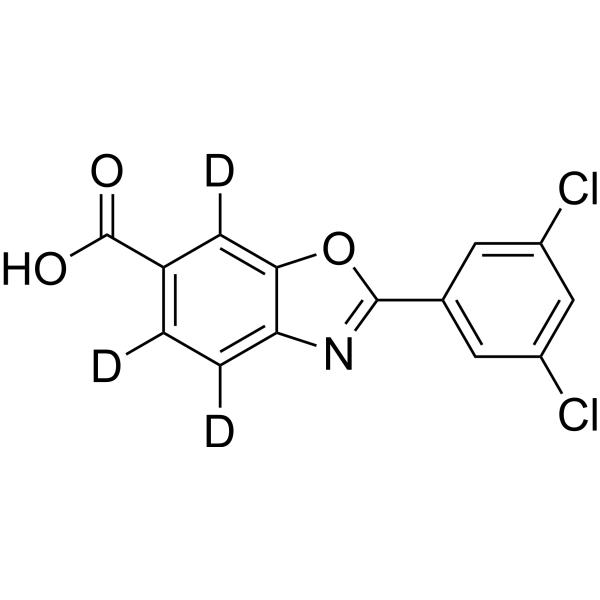
Tafamidis-d3 is deuterium labeled Tafamidis. Tafamidis is a potent and selective transthyretin (TTR) stabilizer, shows comparable potency and efficacy to the mutumant homotetramers V30M-TTR, V122I-TTR and wild type WT-TTR, with EC50s of 2.7-3.2 μM. Tafamidis inhibits amyloidogenesis[1].
PROTAC BRAF-V600E degrader-2 (compound 12) is a potent BRAF-V600E degrader with Kds of 14.4 nM and 9.5 nM for BRAF and BRAF-V600E, respectively. PROTAC BRAF-V600E degrader-2 selectively degraded the kinase domain of BRAF-V600E but not the wild-type BRAF. PROTAC BRAF-V600E degrader-2 inhibits melanoma cell growth .
Bafilomycin D is a specific inhibitor of vacuolar-type ATPase (V-ATPase). Bafilomycin D has antimicrobial, insecticidal, herbicidal and cytotoxic activity .
FKBP51F67V-selective antagonist Ligand2 (example 3-3) is a potent FKBP51 F67V-selective antagonist ligand. FKBP51F67V-selective antagonist Ligand2 reverses the anxiogenic phenotype induced by overexpression of FKBP51 F67V in the amygdala. FKBP51F67V-selective antagonist Ligand2 binds to FKBP51 F67V, but not to wild-type FKBP51 or FKBP52 .
ML336 is quinazolinone-based inhibitor against venezuelan equine encephalitis virus (VEEV), with IC50s of 32, 20, and 42 nM for VEEV TC-83 CPE , VEEV V3526 CPE, VEEV Wild Type CPE, respectively. ML336 potently inhibits a VEEV-induced cytopathic effect in three strains of the virus (TC-83, V3526, and wild type Trinidad donkey) in the low nanomolar range .
PLX-4720 is a potent and selective inhibitor of B-Raf V600E with IC50 of 13 nM in a cell-free assay, equally potent to c-Raf-1(Y340D and Y341D mutations), and 10-fold selectivity for B-Raf V600E than wild-type B-Raf.
Barbadin is a novel and selective β-arrestin/β2-adaptin interaction inhibitor, has IC50 values of 19.1 μM for β-arrestin1 and 15.6 μM for β-arrestin2. Barbadin blocks agonist-promoted endocytosis of the prototypical β2-adrenergic, V2-vasopressin and angiotensin-II type-1 receptors. Barbadin can induce apoptosis .
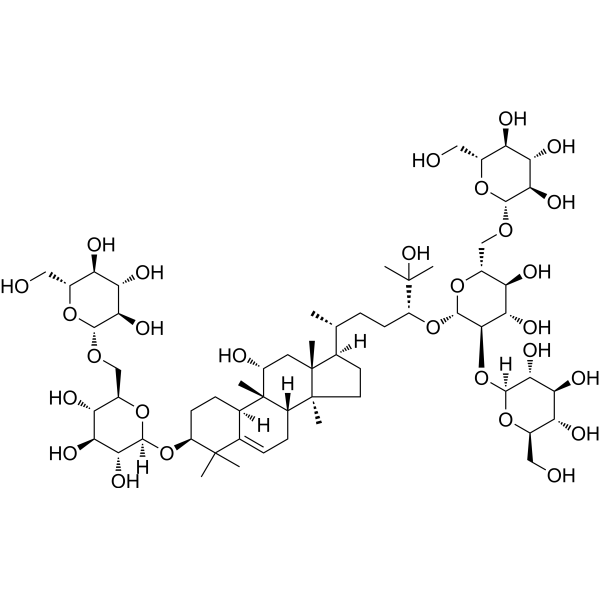
Mogroside V is a the major active constituent of a traditional Chinese medicine Siraitiae Fructus. Mogroside V reduces the intracellular reactive oxygen species (ROS) levels and enhances mitochondrial function. Mogroside V has anti-oxidative, anti-diabetic and anti-carcinogenic effects. Mogroside V can be used for diabetic diseases research .
sPLA2-X Inhibitor 31 is a selective secreted phospholipase A2type X (sPLA2-X) inhibitor with IC50s of 26 nM, 310 nM, and 2230 nM for sPLA2-X, sPLA2-IIa, and sPLA2-V, respectively .
FGFR-IN-8 (Compound 17a) is a highly potent and orally active panFGFR inhibitor against wild-type and mutant FGFRs. FGFR-IN-8 shows inhibition with IC50 values of <0.5, 189.1, <0.5, 22.6, <0.5 and 7.30 nM against FGFR1, V564F-FGFR2, N549H-FGFR2, V555M-FGFR3, FGFR3 and FGFR4, respectively. GFR-IN-8 induces cancer cell apoptosis and shows anticancer activities .
Futibatinib (TAS-120) is an orally bioavailable, highly selective, and irreversible FGFR inhibitor, with IC50s of 3.9, 1.3, 1.6, and 8.3 nM for FGFR 1-4, respectively. Futibatinib inhibits mutant and wild-type FGFR2 with similar IC50s (wild-type FGFR2=0.9 nM; V5651=1-3 nM; N550H=3.6 nM; E566G=2.4 nM) .
Concanamycin A (Folimycin; Antibiotic X 4357B) is a macrolide antibiotic, a vacuolar type H +-ATPase (V-ATPase) inhibitor. Concanamycin A is also an inhibitor of lysosomal acidification, can be used to T cell-mediated inflammation research - .
c-Met-IN-18 is ATP competitive type-III c-MET inhibitor of WT and D1228V mutant c-MET. c-Met-IN-18 has inhibitory for WT/D1228V with an IC50 value of 0.013/0.20 e.c-Met-IN-18 can be used for the research of c-MET driven cancers . c-Met-IN-18 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Gcase activator 2 (compound 14), a pyrrolo[2,3-b]pyrazine, is alos a β-Glucocerebrosidase (GCase) activator (EC50=3.8 μM). Gcase activator 2 induces GCase dimerizatio (both K-type and V-type). And Gcase activator 2 has low metabolic clearance in human and mouse .
AMG0347 is a transient receptor potential typeV1 receptor antagonist. AMG0347 inhibits activation of the rat TRPV1 channel by heat (IC50 = 0.2 nm), protons (IC50= 0.8 nm), or capsaicin (IC50 = 0.7 nm) .
Bcl-2-IN-5 is a BCL-2 inhibitor with IC50s of 0.12 nM, 0.14 nM and 0.22 nM for Bcl-2 wild type, Bcl-2 D103Y and Bcl-2 G101V, respectively. Bcl-2-IN-5 inhibits the cell growth with IC50 values of 0.2 nM and 0.44 nM for Bcl 2-G101V knock-in RS4; 11 and RS4; 11 cells, respectively (WO2021208963A1; Example 155) .
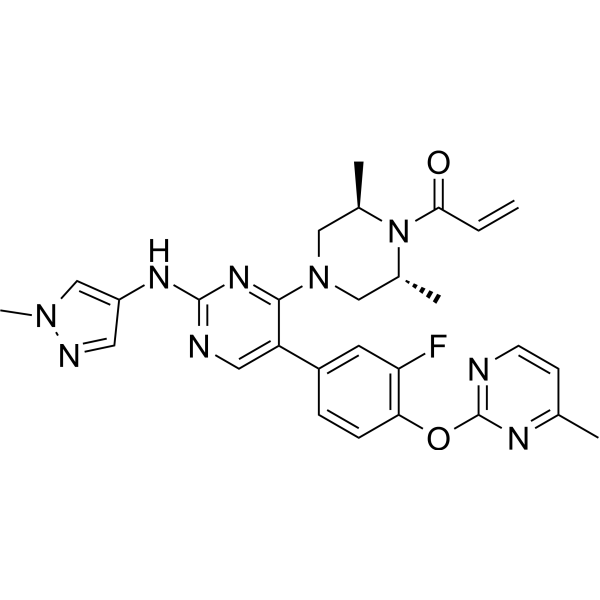
Zeteletinib (BOS-172738; DS-5010) is an orally active, selective RET kinase inhibitor with nanomolar potency against RET and >300-fold selectivity against VEGFR2. Zeteletinib shows exquisite potency for the wild typeRET, RET V804M/L gatekeeper mutants, and the most common oncogenic RET mutation M918T. Zeteletinib has potent antitumor activity .
Vem-L-Cy5 (compound 3),modified with the NIR fluorophore cyanine-5 (Cy5),is a Vemurafenib (HY-12057)-based inhibitor of BRAF. Vem-L-Cy5 targets to BRAF V600E,and also inhibits MEK phosphorylation. Vem-L-Cy5 has cell permeability,and inhibits cell growth of many types of cancer .
AZ304 is an ATP-competitive dual BRAF kinase inhibitor, potently inhibits wild type BRAF, V600E mutant BRAF and wild type CRAF, with IC50s of 79 nM, 38 nM and 68 nM, respectively. AZ304 also has significant effect on other kinases, such as p38 (IC50, 6 nM), CSF1R (IC50, 35 nM). Anti-tumor activity .
Zeteletinib (BOS-172738; DS-5010) hemiadipate is an orally active, selective RET kinase inhibitor with nanomolar potency against RET and >300-fold selectivity against VEGFR2. Zeteletinib hemiadipate shows exquisite potency for the wild typeRET, RET V804M/L gatekeeper mutants, and the most common oncogenic RET mutation M918T. Zeteletinib hemiadipate has potent antitumor activity .
CHMFL-ABL-039 is a type II native ABL kinase and drug-resistant V299L mutant BCR-ABL inhibitor with the IC50s of 7.9 nM and 27.9 nM, respectively. CHMFL-ABL-039 is used in the research of chronic myeloid leukemia .
FGFR4-IN-8 (Compound 7v) is an ATP-competitive, highly selective covalent inhibitor of wild-type and gatekeeper mutant FGFR4. FGFR4-IN-8 exhibits excellent potency against FGFR4, FGFR4 V550L, FGFR4 V550M and FGFR4 C552S with IC50s of 0.5, 0.25, 1.6, 931 nM, respectively. FGFR4-IN-8 exhibits potent antiproliferative activity against Hep3B hepatocellular carcinoma cells with the IC50 value of 29 nM. FGFR4-IN-8 demonstrates modest in vivo antitumor efficacy in nude mice bearing the Huh-7 xenograft model .
L-372662 is a potent and orally active non-peptide oxytocin antagonist with a Ki value of 4.8. The Kd value of L-372662 for wild-type hOTR and [A318G]OTR is 5.8 nM and 73 nM. L-372662 shows selectivity to OTR:V1aR .
Pacritinib (SB1518) is a potent inhibitor of both wild-typeJAK2 (IC50=23 nM) and JAK2 V617F mutant (IC50=19 nM). Pacritinib also inhibits FLT3 (IC50=22 nM) and its mutant FLT3 D835Y (IC50=6 nM).
Bafilomycin C1 is a macrolide antibiotic isolated from Streptomyces sp. Bafilomycin C1 is a potent, specific and reversible inhibitor of vacuolar-type H +-ATPases (V-ATPases). Bafilomycin C1 inhibits growth of gram-positive bacteria and fungi . Bafilomycin C1 induces cell apoptosis and can be used for the study of hepatocellular carcinoma (HCC) .
ω-Conotoxin FVIA is an N-type Ca 2+channel (Cav2.2)
inhibitor. ω-Conotoxin FVIA reduces mechanical and thermal pain abnormalities
in a rat model of caudal nerve injury. ω-Conotoxin FVIA can be used in the
study of highly effective pain relievers with low side effects
.
Talacotuzumab (JNJ 56022473; CSL 362) is an IgG1-type fully humanized, CD123-neutralizing monoclonal antibody containing a modified Fc structure. Talacotuzumab has KDs of 0.43 nM, 188 nM, 46 nM, 16.8 nM for CD123, CD32b/c, CD16-158F, CD16-158V, respectively. Talacotuzumab inhibits IL-3 binding to CD123, antagonizing IL-3 signaling in target cells. Talacotuzumab has mutated the Fc region to increase affinity for CD16 (FcγRIIIa), thereby enhancing antibody-dependent cell-mediated cytotoxicity (ADCC). Talacotuzumab is highly effective in vivo reducing leukemic cell growth in acute myeloid leukemia (AML) xenograft mouse models .
RET-IN-22 (compound 17b) is a potent, selective and orally active RET inhibitor with an IC50 of 20.9 nM and 18.3 nM for wild-type RET and RET-V804M, respectively. RET-IN-22 shows highly selective profile to most kinases, especially to EGFR and VEGFR2. RET-IN-22 has anticancer effects .
IHMT-EZH2-426 (compound 38) is a potent and covalent EZH2 degrader with IC50s of 1.3 nM, 1.2 nM, and 1.7-3.5 nM against EZH2 wild-type, EZH2-A687V, and EZH2-Y641F/Y641N/Y641S, respectively. IHMT-EZH2-426 exhibits potent antiproliferation effects against both B-cell lymphoma and triple negative breast cancer (TNBC) cell lines by reducing the levels of H3K27me3 and EZH2 .
Diphyllin is an arylnaphthalene lignan isolated from Justicia procumbens and is a potent HIV-1 inhibitor with an IC50 of 0.38 μM. Diphyllin is active against vesicular stomatitis virus (VSV) and influenza virus . Diphyllin is a vacuolar type H +-ATPase (V-ATPase) inhibitor with an IC50 value of 17 nM and inhibits lysosomal acidification in human osteoclasts . Diphyllin inhibits NO production with an IC50 of 50 μM and has anticancer and anti-inflammatory activities .
Pacritinib hydrochloride is a potent inhibitor of both wild-typeJAK2 (IC50=23 nM) and JAK2 V617F mutant (IC50=19 nM). Pacritinib hydrochloride also inhibits FLT3 (IC50=22 nM) and its mutant FLT3 D835Y (IC50=6 nM). Pacritinib hydrochloride can be used for the research of acute myeloid leukemia (AML) and myelofibrosis (MF) .
Pacritinib (SB1518) citrate is a potent inhibitor of both wild-typeJAK2 (IC50=23 nM) and JAK2 V617F mutant (IC50=19 nM). Pacritinib citrate also inhibits FLT3 (IC50=22 nM) and its mutant FLT3 D835Y (IC50=6 nM). Pacritinib citrate can be used for the research of acute myeloid leukemia (AML) and myelofibrosis (MF) .
BSc5371 is a potent and irreversible FLT3 inhibitor, with Kds of 1.3, 0.83, 1.5, 5.8 and 2.3 nM for mutant FLT3(D835H), FLT3(ITD, D835V), FLT3(ITD, F691L), FLT3-ITD and wild type FLT3wt, respectively. BSc5371 is cytotoxic to FLT3-dependent cell lines .
SAH-SOS1A is a peptide-based SOS1/KRAS protein interaction inhibitor. SAH-SOS1A binds to wild-type and mutant KRAS (G12D, G12V, G12C, G12S, and Q61H) with nanomolar affinity (EC50=106-175 nM), directly and independently blocks nucleotide association, impairs KRAS-driven cancer cell viability, and exerts its effects by on-mechanism blockade of the ERK-MAPK phosphosignaling cascade downstream of KRAS .
EZH2-IN-9 is a potent inhibitor of EZH2. EZH2 overexpression or mutations in the SET region (Y641F, Y641N, A687V, A677G point mutations) all lead to abnormal elevation of H3K27me3 and promote the growth and development of many types of tumors, such as breast cancer, prostate cancer, leukemia, etc. EZH2-IN-9 has the potential for the research of cancer diseases (extracted from patent WO2021180235A1, compound 17) .
EZH2-IN-7 is a potent inhibitor of EZH2. EZH2 overexpression or mutations in the SET region (Y641F, Y641N, A687V, A677G point mutations) all lead to abnormal elevation of H3K27me3 and promote the growth and development of many types of tumors, such as breast cancer, prostate cancer, leukemia, etc. EZH2-IN-7 has the potential for the research of cancer diseases (extracted from patent WO2021129629A1, compound 259) .
FGFR-IN-10 is an orally active inhibitor of FGFR and Cytochrome P450 (CYPs). FGFR-IN-10 inhibits wide type and V564F mutant FGFR2 with IC50s of 104.1 nM and 43.6 nM, respectively. FGFR-IN-10 also inhibits CYPs with IC50s of 3.33 μM (CYP2C9), 18.75 μM (CYP2C19), 4.34 μM (CYP2CD6), and 0.69 μM (CYP3A4), respectively .
XL019 is a potent, orally active, and selective JAK2 inhibitor, with IC50s of 2.2, 134.3, and 214.2 nM for JAK2, JAK1 and JAK3, respectively. XL019 shows 50-fold or greater selectivity for JAK2, versus a panel of over 100 serine/threonine and tyrosine kinases, including other members of the JAK family. XL019 potently inhibits STAT3 and STAT5 phosphorylation in cells harboring either JAK2V617F or wild-type JAK2 .
SAH-SOS1A TFA is a peptide-based SOS1/KRAS protein interaction inhibitor. SAH-SOS1A TFA binds to wild-type and mutant KRAS (G12D, G12V, G12C, G12S, and Q61H) with nanomolar affinity (EC50=106-175 nM). SAH-SOS1A TFA directly and independently blocks nucleotide association. SAH-SOS1A TFA impairs KRAS-driven cancer cell viability and exerts its effects by on-mechanism blockade of the ERK-MAPK phosphosignaling cascade downstream of KRAS .
G12 (Ras 5-17) is a wild-type Ras peptide consisted of amino acids 5-17 (KLVVVGAGGVGKS). G12 can be used as a control of mutant Ras peptides studies (such V12) .
SAH-SOS1A TFA is a peptide-based SOS1/KRAS protein interaction inhibitor. SAH-SOS1A TFA binds to wild-type and mutant KRAS (G12D, G12V, G12C, G12S, and Q61H) with nanomolar affinity (EC50=106-175 nM). SAH-SOS1A TFA directly and independently blocks nucleotide association. SAH-SOS1A TFA impairs KRAS-driven cancer cell viability and exerts its effects by on-mechanism blockade of the ERK-MAPK phosphosignaling cascade downstream of KRAS .
G12 (Ras 5-17) TFA is a wild-type Ras peptide consisted of amino acids 5-17 (KLVVVGAGGVGKS). G12 TFA can be used as a control of mutant Ras peptides studies (such V12) .
ω-Conotoxin FVIA is an N-type Ca 2+channel (Cav2.2)
inhibitor. ω-Conotoxin FVIA reduces mechanical and thermal pain abnormalities
in a rat model of caudal nerve injury. ω-Conotoxin FVIA can be used in the
study of highly effective pain relievers with low side effects
.
SAH-SOS1A is a peptide-based SOS1/KRAS protein interaction inhibitor. SAH-SOS1A binds to wild-type and mutant KRAS (G12D, G12V, G12C, G12S, and Q61H) with nanomolar affinity (EC50=106-175 nM), directly and independently blocks nucleotide association, impairs KRAS-driven cancer cell viability, and exerts its effects by on-mechanism blockade of the ERK-MAPK phosphosignaling cascade downstream of KRAS .
Talacotuzumab (JNJ 56022473; CSL 362) is an IgG1-type fully humanized, CD123-neutralizing monoclonal antibody containing a modified Fc structure. Talacotuzumab has KDs of 0.43 nM, 188 nM, 46 nM, 16.8 nM for CD123, CD32b/c, CD16-158F, CD16-158V, respectively. Talacotuzumab inhibits IL-3 binding to CD123, antagonizing IL-3 signaling in target cells. Talacotuzumab has mutated the Fc region to increase affinity for CD16 (FcγRIIIa), thereby enhancing antibody-dependent cell-mediated cytotoxicity (ADCC). Talacotuzumab is highly effective in vivo reducing leukemic cell growth in acute myeloid leukemia (AML) xenograft mouse models .
Mogroside V is a the major active constituent of a traditional Chinese medicine Siraitiae Fructus. Mogroside V reduces the intracellular reactive oxygen species (ROS) levels and enhances mitochondrial function. Mogroside V has anti-oxidative, anti-diabetic and anti-carcinogenic effects. Mogroside V can be used for diabetic diseases research .
Bafilomycin C1 is a macrolide antibiotic isolated from Streptomyces sp. Bafilomycin C1 is a potent, specific and reversible inhibitor of vacuolar-type H +-ATPases (V-ATPases). Bafilomycin C1 inhibits growth of gram-positive bacteria and fungi . Bafilomycin C1 induces cell apoptosis and can be used for the study of hepatocellular carcinoma (HCC) .
Diphyllin is an arylnaphthalene lignan isolated from Justicia procumbens and is a potent HIV-1 inhibitor with an IC50 of 0.38 μM. Diphyllin is active against vesicular stomatitis virus (VSV) and influenza virus . Diphyllin is a vacuolar type H +-ATPase (V-ATPase) inhibitor with an IC50 value of 17 nM and inhibits lysosomal acidification in human osteoclasts . Diphyllin inhibits NO production with an IC50 of 50 μM and has anticancer and anti-inflammatory activities .
Bafilomycin D is a specific inhibitor of vacuolar-type ATPase (V-ATPase). Bafilomycin D has antimicrobial, insecticidal, herbicidal and cytotoxic activity .
ATP6V1F is an important subunit of the vacuolar (H+)-ATPase (V-ATPase) V1 complex and is an important component of this enzyme's role in cellular pH regulation. It forms a catalytic AB heterodimer and peripheral stem that plays a role in ATP hydrolysis. ATP6V1F Protein, Human is the recombinant human-derived ATP6V1F protein, expressed by E. coli , with tag free. The total length of ATP6V1F Protein, Human is 119 a.a., with molecular weight of ~13 KDa.
ATP6V1F is an important subunit of the vacuolar (H+)-ATPase (V-ATPase) V1 complex and is an important component of this enzyme's role in cellular pH regulation. It forms a catalytic AB heterodimer and peripheral stem that plays a role in ATP hydrolysis. ATP6V1F Protein, Human (GST) is the recombinant human-derived ATP6V1F protein, expressed by E. coli , with N-GST labeled tag. The total length of ATP6V1F Protein, Human (GST) is 119 a.a., with molecular weight of ~40 KDa.
RNASEK proteins cleave ApU and ApG phosphodiester bonds and hydrolyze UpU bonds at a slower rate. RNASEK Protein, Human (Cell-Free, His) is the recombinant human-derived RNASEK protein, expressed by E. coli Cell-free , with N-10*His labeled tag. The total length of RNASEK Protein, Human (Cell-Free, His) is 137 a.a., with molecular weight of 21.5 kDa.
MARCHF5 protein is a mitochondrial E3 ubiquitin protein ligase that actively regulates mitochondrial fission and affects mitochondrial morphology. Additionally, it plays a key role in preventing cellular senescence and aids in mitochondrial quality control. MARCHF5 Protein, Mouse (Cell-Free, His) is the recombinant mouse-derived MARCHF5 protein, expressed by E. coli Cell-free , with N-6*His labeled tag. The total length of MARCHF5 Protein, Mouse (Cell-Free, His) is 278 a.a., with molecular weight of 32.2 kDa.
MARCHF5 protein is a mitochondrial E3 ubiquitin protein ligase that actively regulates mitochondrial fission and affects mitochondrial morphology. Additionally, it plays a key role in preventing cellular senescence and aids in mitochondrial quality control. MARCHF5 Protein, Mouse (Cell-Free, His, HiBiT) is the recombinant mouse-derived MARCHF5 protein, expressed by E. coli Cell-free , with N-HiBiT, N-8*His labeled tag. The total length of MARCHF5 Protein, Mouse (Cell-Free, His, HiBiT) is 278 a.a., with molecular weight of 35.6 kDa.
VISTA/B7-H5 Protein is a type I transmembrane protein expressed primarily in white blood cells that inhibits T cell function. VISTA/B7-H5 Protein promotes embryonic stem cell differentiation by inhibiting BMP4 signaling and stimulates MMP14 mediated MMP2 activation. VISTA/B7-H5 Protein is highly expressed in tumors. VISTA/B7-H5 Protein, Mouse (159a.a, HEK293, His) is the recombinant mouse-derived VISTA/B7-H5 protein, expressed by HEK293 , with C-6*His labeled tag. The total length of VISTA/B7-H5 Protein, Mouse (159a.a, HEK293, His) is 159 a.a., with molecular weight of 30-40 kDa.
The PRDX5 protein (or Peroxiredoxin-5) plays a critical role as a thiol-specific peroxidase that reduces hydrogen peroxide and organic hydroperoxides. This enzyme activity is essential for cells to defend against oxidative stress and detoxify peroxides. PRDX5/Peroxiredoxin-5 Protein, Human (HEK293, His) is the recombinant human-derived PRDX5/Peroxiredoxin-5 protein, expressed by HEK293 , with N-6*His labeled tag. The total length of PRDX5/Peroxiredoxin-5 Protein, Human (HEK293, His) is 162 a.a., with molecular weight of ~17.0 kDa.
The PCSK9 protein is an important regulator of plasma cholesterol homeostasis, affecting LDL receptor family members such as LDLR and VLDLR. It promotes their intracellular degradation and enhances hepatic LDLR degradation through non-proteolytic mechanisms. PCSK9 Protein, Human (HEK293, V474I, G670E, His) is the recombinant human-derived PCSK9 protein, expressed by HEK293 , with C-6*His labeled tag and V474I, G670E, , , mutation. The total length of PCSK9 Protein, Human (HEK293, V474I, G670E, His) is 662 a.a., with molecular weight of 19 & 60 kDa, respectively.
The PCSK9 protein is an important regulator of plasma cholesterol homeostasis, affecting LDL receptor family members such as LDLR and VLDLR. It promotes their intracellular degradation and enhances hepatic LDLR degradation through non-proteolytic mechanisms. PCSK9 Protein, Human (Biotinylated, V474I, G670E, HEK293, His-HA-Avi) is the recombinant human-derived PCSK9 protein, expressed by HEK293 , with C-Avi, C-HA, C-8*His labeled tag and V474I, G670E, , , mutation. The total length of PCSK9 Protein, Human (Biotinylated, V474I, G670E, HEK293, His-HA-Avi) is 662 a.a., with molecular weight of 18 & 58-70 & 90-150 kDa, respectively.
Tafamidis-d3 is deuterium labeled Tafamidis. Tafamidis is a potent and selective transthyretin (TTR) stabilizer, shows comparable potency and efficacy to the mutumant homotetramers V30M-TTR, V122I-TTR and wild type WT-TTR, with EC50s of 2.7-3.2 μM. Tafamidis inhibits amyloidogenesis[1].
c-Met-IN-18 is ATP competitive type-III c-MET inhibitor of WT and D1228V mutant c-MET. c-Met-IN-18 has inhibitory for WT/D1228V with an IC50 value of 0.013/0.20 e.c-Met-IN-18 can be used for the research of c-MET driven cancers . c-Met-IN-18 is a click chemistry reagent, it contains an Alkyne group and can undergo copper-catalyzed azide-alkyne cycloaddition (CuAAc) with molecules containing Azide groups.
Inquiry Online
Your information is safe with us. * Required Fields.